Nernst equation
In electrochemistry, the Nernst equation is an equation that can be used (in conjunction with other information) to determine the equilibrium reduction potential of a half-cell in an electrochemical cell. It can also be used to determine the total voltage (electromotive force) for a full electrochemical cell. It is named after the German physical chemist who first formulated it, Walther Nernst.[1][2]
Contents |
Expression
The two (ultimately equivalent) equations for these two cases (half-cell, full cell) are as follows:
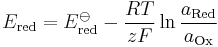 (half-cell reduction potential)
(half-cell reduction potential)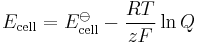 (total cell potential)
(total cell potential)
where
- Ered is the half-cell reduction potential at the temperature of interest
- E
ored is the standard half-cell reduction potential - Ecell is the cell potential (electromotive force)
- E
ocell is the standard cell potential at the temperature of interest - R is the universal gas constant: R = 8.314 472(15) J K−1 mol−1
- T is the absolute temperature
- a is the chemical activity for the relevant species, where aRed is the reductant and aOx is the oxidant. aX = γXcX, where γX is the activity coefficient of species X. (Since activity coefficients tend to unity at low concentrations, activities in the Nernst equation are frequently replaced by simple concentrations.)
- F is the Faraday constant, the number of coulombs per mole of electrons: F = 9.648 533 99(24)×104 C mol−1
- z is the number of moles of electrons transferred in the cell reaction or half-reaction
- Q is the reaction quotient.
At room temperature (25 °C), RT/F may be treated like a constant and replaced by 25.693 mV for cells.
The Nernst equation is frequently expressed in terms of base 10 logarithms (i.e., common logarithms) rather than natural logarithms, in which case it is written, for a cell at 25 °C:
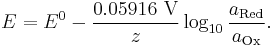
The Nernst equation is used in physiology for finding the electric potential of a cell membrane with respect to one type of ion.
Nernst potential
The Nernst equation has a physiological application when used to calculate the potential of an ion of charge z across a membrane. This potential is determined using the concentration of the ion both inside and outside the cell:
![E = \frac{R T}{z F} \ln\frac{[\text{ion outside cell}]}{[\text{ion inside cell}]} = 2.303\frac{R T}{z F} \log_{10}\frac{[\text{ion outside cell}]}{[\text{ion inside cell}]}.](/2012-wikipedia_en_all_nopic_01_2012/I/4c767eef648cf4f2007111d25fe5921d.png)
When the membrane is in thermodynamic equilibrium (i.e., no net flux of ions), the membrane potential must be equal to the Nernst potential. However, in physiology, due to active ion pumps, the inside and outside of a cell are not in equilibrium. In this case, the resting potential can be determined from the Goldman equation:
![E_{m} = \frac{RT}{F} \ln{ \left( \frac{ \sum_{i}^{N} P_{M^{%2B}_{i}}[M^{%2B}_{i}]_\mathrm{out} %2B \sum_{j}^{M} P_{A^{-}_{j}}[A^{-}_{j}]_\mathrm{in}}{ \sum_{i}^{N} P_{M^{%2B}_{i}}[M^{%2B}_{i}]_\mathrm{in} %2B \sum_{j}^{M} P_{A^{-}_{j}}[A^{-}_{j}]_\mathrm{out}} \right) }](/2012-wikipedia_en_all_nopic_01_2012/I/b484cb90615da3a7b33eef659f2da93e.png)
 = The membrane potential (in volts, equivalent to joules per coulomb)
= The membrane potential (in volts, equivalent to joules per coulomb)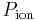 = the permeability for that ion (in meters per second)
= the permeability for that ion (in meters per second)![[ion]_\mathrm{out}](/2012-wikipedia_en_all_nopic_01_2012/I/dc7e9cedbf6b51c90997a1108fa6251b.png) = the extracellular concentration of that ion (in moles per cubic meter, to match the other SI units, though the units strictly don't matter, as the ion concentration terms become a dimensionless ratio)
= the extracellular concentration of that ion (in moles per cubic meter, to match the other SI units, though the units strictly don't matter, as the ion concentration terms become a dimensionless ratio)![[ion]_\mathrm{in}](/2012-wikipedia_en_all_nopic_01_2012/I/d4398d66174b0fca2224cd19631589bc.png) = the intracellular concentration of that ion (in moles per cubic meter)
= the intracellular concentration of that ion (in moles per cubic meter)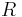 = The ideal gas constant (joules per kelvin per mole)
= The ideal gas constant (joules per kelvin per mole)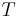 = The temperature in kelvin
= The temperature in kelvin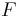 = Faraday's constant (coulombs per mole)
= Faraday's constant (coulombs per mole)
The potential across the cell membrane that exactly opposes net diffusion of a particular ion through the membrane is called the Nernst potential for that ion. As seen above, the magnitude of the Nernst potential is determined by the ratio of the concentrations of that specific ion on the two sides of the membrane. The greater this ratio the greater the tendency for the ion to diffuse in one direction, and therefore the greater the Nernst potential required to prevent the diffusion.
Derivation
Using Boltzmann factors
For simplicity, we will consider a solution of redox-active molecules that undergo a one-electron reversible reaction
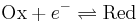
and that have a standard potential of zero. The chemical potential 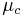 of this solution is the difference between the energy barriers for taking electrons from and for giving electrons to the working electrode that is setting the solution's electrochemical potential.
of this solution is the difference between the energy barriers for taking electrons from and for giving electrons to the working electrode that is setting the solution's electrochemical potential.
The ratio of oxidized to reduced molecules, [Ox]/[Red], is equivalent to the probability of being oxidized (giving electrons) over the probability of being reduced (taking electrons), which we can write in terms of the Boltzmann factors for these processes:
![\frac{[\mathrm{Ox}]}{[\mathrm{Red}]}
= \frac{\exp \left(-[\mbox{barrier for losing an electron}]/kT\right)}
{\exp \left(-[\mbox{barrier for gaining an electron}]/kT\right)}
= \exp \left(\mu_c / kT \right).](/2012-wikipedia_en_all_nopic_01_2012/I/cced0eb2349e0406bfdbd755b1605c1a.png)
Taking the natural logarithm of both sides gives
![\mu_c = kT \ln \frac{[\mathrm{Ox}]}{[\mathrm{Red}]}.](/2012-wikipedia_en_all_nopic_01_2012/I/ff5f0da2623e3f6c10e3e28de6c1bdd6.png)
If 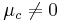 at [Ox]/[Red] = 1, we need to add in this additional constant:
at [Ox]/[Red] = 1, we need to add in this additional constant:
![\mu_c = \mu_c^0 %2B kT \ln \frac{[\mathrm{Ox}]}{[\mathrm{Red}]}.](/2012-wikipedia_en_all_nopic_01_2012/I/f64007dc5d671977fa1118f048af0eba.png)
Dividing the equation by e to convert from chemical potentials to electrode potentials, and remembering that kT/e = RT/F, we obtain the Nernst equation for the one-electron process 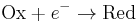 :
:
![E = E^0 %2B \frac{kT}{e} \ln \frac{[\mathrm{Ox}]}{[\mathrm{Red}]}
= E^0 - \frac{RT}{F} \ln \frac{[\mathrm{Red}]}{[\mathrm{Ox}]}.](/2012-wikipedia_en_all_nopic_01_2012/I/ada2c10e0b4876c9638e92e8d7515e16.png)
Using entropy and Gibbs energy
Quantities here are given per molecule, not per mole, and so Boltzmann constant k and the electron charge e are used instead of the gas constant R and Faraday's constant F. To convert to the molar quantities given in most chemistry textbooks, it is simply necessary to multiply by Avogadro's number: 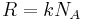 and
and 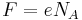 .
.
The entropy of a molecule is defined as
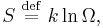
where 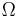 is the number of states available to the molecule. The number of states must vary linearly with the volume V of the system, which is inversely proportional to the concentration c, so we can also write the entropy as
is the number of states available to the molecule. The number of states must vary linearly with the volume V of the system, which is inversely proportional to the concentration c, so we can also write the entropy as
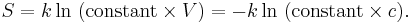
The change in entropy from some state 1 to another state 2 is therefore
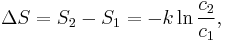
so that the entropy of state 2 is
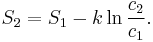
If state 1 is at standard conditions, in which 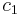 is unity (e.g., 1 atm or 1 M), it will merely cancel the units of
is unity (e.g., 1 atm or 1 M), it will merely cancel the units of 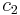 . We can, therefore, write the entropy of an arbitrary molecule A as
. We can, therefore, write the entropy of an arbitrary molecule A as
![S(A) = S^0(A) - k \ln [A], \,](/2012-wikipedia_en_all_nopic_01_2012/I/98a583e9be5c433e5c18c8a8bca71688.png)
where 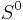 is the entropy at standard conditions and [A] denotes the concentration of A. The change in entropy for a reaction
is the entropy at standard conditions and [A] denotes the concentration of A. The change in entropy for a reaction
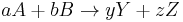
is then given by
![\Delta S_\mathrm{rxn} = [yS(Y) %2B zS(Z)] - [aS(A) %2B bS(B)]
= \Delta S^0_\mathrm{rxn} - k \ln \frac{[Y]^y [Z]^z}{[A]^a [B]^b}.](/2012-wikipedia_en_all_nopic_01_2012/I/ec760f7fa1df0c573b733c2a9d84de53.png)
We define the ratio in the last term as the reaction quotient:
![Q = \frac{\prod_j a_j^{\nu_j}}{\prod_i a_i^{\nu_i}} \approx \frac{[Z]^z [Y]^y}{[A]^a [B]^b}.](/2012-wikipedia_en_all_nopic_01_2012/I/d1123122466ad21807b175ea58ab1c6f.png)
where the numerator is a product of reaction product activities, a j, each raised to the power of a stoichiometric coefficient, ν j, and the denominator is a similar product of reactant activities. All activities refer to a time t. Under certain circumstances (see chemical equilibrium) each activity term such as 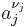 may be replaced by a concentration term, [A]. In an electrochemical cell, the cell potential E is the chemical potential available from redox reactions (
may be replaced by a concentration term, [A]. In an electrochemical cell, the cell potential E is the chemical potential available from redox reactions (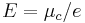 ). E is related to the Gibbs energy change
). E is related to the Gibbs energy change 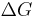 only by a constant:
only by a constant: 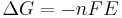 , where n is the number of electrons transferred and is the Faraday constant. There is a negative sign because a spontaneous reaction has a negative free energy and a positive potential E. The Gibbs energy is related to the entropy by
, where n is the number of electrons transferred and is the Faraday constant. There is a negative sign because a spontaneous reaction has a negative free energy and a positive potential E. The Gibbs energy is related to the entropy by 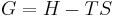 , where H is the enthalpy and T is the temperature of the system. Using these relations, we can now write the change in Gibbs energy,
, where H is the enthalpy and T is the temperature of the system. Using these relations, we can now write the change in Gibbs energy,
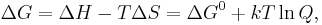
and the cell potential,
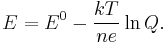
This is the more general form of the Nernst equation. For the redox reaction 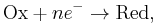
![Q = \frac{[\mathrm{Red}]}{[\mathrm{Ox}]}](/2012-wikipedia_en_all_nopic_01_2012/I/a6a90b70137e2c472f19048ab974d7b1.png) , and we have:
, and we have:
![E = E^0 - \frac{kT}{ne} \ln \frac{[\mathrm{Red}]}{[\mathrm{Ox}]}
= E^0 - \frac{RT}{nF} \ln \frac{[\mathrm{Red}]}{[\mathrm{Ox}]}
= E^0 - \frac{RT}{nF} \ln Q.](/2012-wikipedia_en_all_nopic_01_2012/I/f52e91a64ac9cab618a36fd506a40ca9.png)
The cell potential at standard conditions 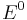 is often replaced by the formal potential
is often replaced by the formal potential 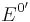 , which includes some small corrections to the logarithm and is the potential that is actually measured in an electrochemical cell.
, which includes some small corrections to the logarithm and is the potential that is actually measured in an electrochemical cell.
Relation to equilibrium
At equilibrium, E = 0 and Q = K. Therefore
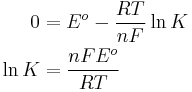
Or at standard temperature,
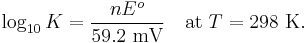
We have thus related the standard electrode potential and the equilibrium constant of a redox reaction.
Limitations
In dilute solutions, the Nernst equation can be expressed directly in terms of concentrations (since activity coefficients are close to unity). But at higher concentrations, the true activities of the ions must be used. This complicates the use of the Nernst equation, since estimation of non-ideal activities of ions generally requires experimental measurements.
The Nernst equation also only applies when there is no net current flow through the electrode. The activity of ions at the electrode surface changes when there is current flow, and there are additional overpotential and resistive loss terms which contribute to the measured potential.
At very low concentrations of the potential-determining ions, the potential predicted by Nernst equation approaches toward ±∞. This is physically meaningless because, under such conditions, the exchange current density becomes very low, and then other effects tend to take control of the electrochemical behavior of the system.